
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()


Our research interests center on structural and functional characterization of proteins, genome-wide identification of protein-protein interaction, systematic & quantitative analysis of molecular evolution and biological network for genotype-phenotype mapping of human diseases.
We are developing computational biology methods and bioinformatics tools for network medicine and healthcare. Proteins are the major player of cellular function and they carry out their functional role through complex network of protein-protein interactions. The protein repertoire varies depending on cellular states, tissue type, species, and disease state. However, little is known about how this repertoire changes under different cellular or disease states. To gain a better understanding of these dynamic changes, Kim’s lab is developing essential applications for network biology and large-scale high-throughput data integration analysis. Systemic analysis of protein functional network will provide a framework for understanding how protein compositions respond to changes in human disease states.

I. Disease, medical informatics, cancer big data
Medical bioinformatics
Identification of disease-associated genes, otherwise known as biomarkers, is crucial for early diagnosis or treatment of a disease. Although detection of biomarkers have become less difficult thanks to major improvements in high-throughput sequencing techniques, uncovering biomarkers for polygenic diseases, such as cancers, still remains to be an extremely difficult task due to the intrinsic complexity of the molecular mechanism of the diseases. Therefore, computational methods that handle complex biological problems by combining prior biological knowledge and mathematical models are urgently required to help improve disease treatments. Dr. Kim’s research group tries to provide plausible molecular mechanisms and candidate drug targets by integrating various approaches and large omics databases. Our methods include modeling physical property of protein-protein interactions (PPIs) to understand molecular mechanism of a specific disease, or analyzing large-scale clinical and drug response data that are publically available to make mathematical prediction models.
Molecular subtyping and drug treatment prediction of bladder cancer patients
Muscle-invasive bladder cancer (MIBC) is 9th most common cancer in United States. While the main treatment option for MIBC is cisplatin-based chemotherapy, this drug is known to be only effective for 30% of MIBC patients. Therefore, development of novel drug treatment options is urgently required. Dr Kim’s group is establishing a computational method to first, predict drugs that would be effective for treating MIBC patients and second, identify a subgroup of bladder cancer patients who would benefit most from our predicted drugs.
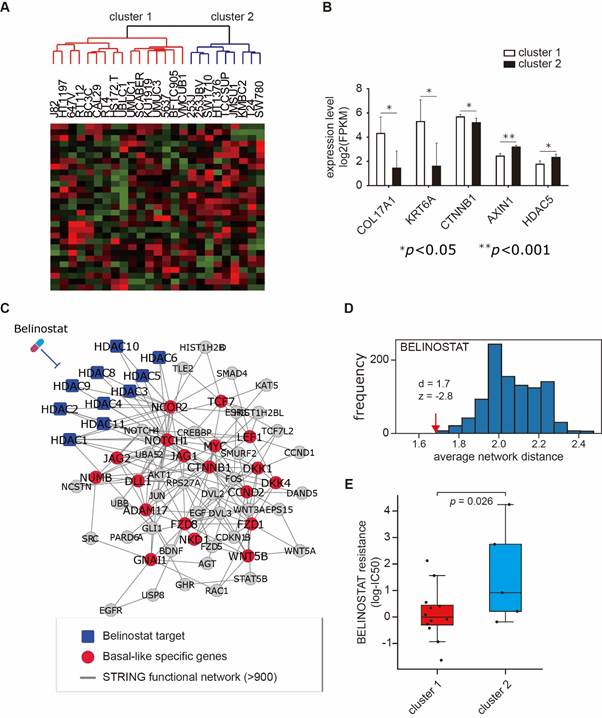
Figure 1. Molecular subtyping of muscle-invasive bladder cancer (MIBC) cell lines (A,B) and prediction and validation of candidate drug to treat MIBC (C-E)
II. Evolutionary analysis of protein sequences
Bioinformatics approaches for the prediction of functionally important sites
Proteins are functional units to play important roles in the biology of the cell. While the number of sequenced genomes continues to increase, experimentally verified functional annotations of whole genomes remains largely unknown. Because experimental investigation is costly and time-consuming, accurate computational methods for predicting protein functions and structures are becoming attractive. Dr. Kim’s research group develops various computational methods for identifying functionally important residues by using evolutionary information.
Prediction of mutational impact based on sequence evolution analysis
As sequencing technology is constantly advancing, many nonsynonymous variants are being detected in a number of genome-wide association studies. However, the majority of the detected variants have trivial impacts on altering the functionality of proteins. Therefore, it is important to evaluate the impacts of these variants on human health and disease, because functional consequence may vary between variants. Dr. Kim’s research group develops various computational methods for predicting impact of variants by analyzing sequence evolution.
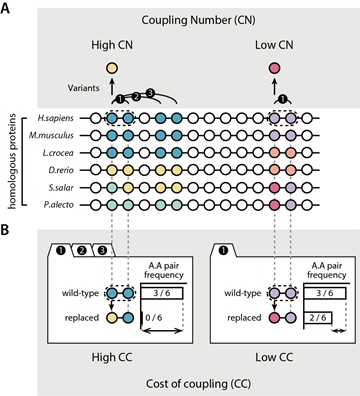
Figure 2. Predicting impacts of variants by analyzing sequence evolution; coupling number and cost of coupling.
III. Protein function and interaction
Protein interaction and network module evolution.
Modular architecture is important for the evolution of cellular systems. Modular rearrangements facilitate functional innovations and modular insulations provide robustness to perturbations. However, molecular-level understanding of the mechanisms underlying modular network evolution is currently not well understood. Through investigating physical property of protein-protein interaction(PPI) in biological modules, we attempt to improve identification of biological modules. Dr.Kim’s research group investigate evolution of modular architecture of PPI network
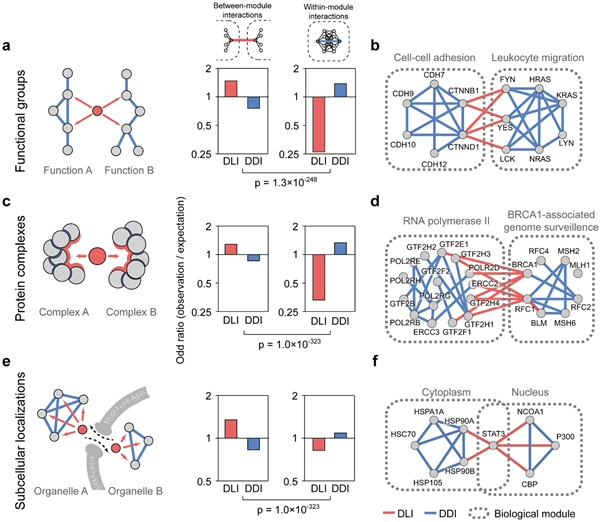
Kim I et al., 2014, PLos Comput Biol.
Figure 3. Enrichment of DLIs and DDIs in interactions between and within biological modules.
Network analysis of high fitness effect gene
Essential genes are those genes of an organism or cell that are thought to be critical for its survival. The centrality-lethality (C-L) rule point out Essential genes (EGs) often form central nodes in protein-protein interaction (PPI) networks. However, many reports have shown that numerous EGs are non-central, suggesting that another principle governs gene essentiality. Using network topology and module analysis in various biological context, we hope to comprehend relationship between vulnerabilities of biological network and gene essentiality.
Protein domain and sub-grouping
Protein functional groups can be categorized by the annotation of protein domains, protein structures, and sequence evolutionary histories. Protein domain is informative to cluster biologically similar proteins, since it is evolutionarily conserved, maintaining its own structure and molecular functions in different proteins within and across species. However, recent studies have reported numerous cases where proteins within a same protein family involved in dissimilar functions. Through, systematic approach of we investigated domain mediated interaction (DMI) information, we develops new measurement of protein community.
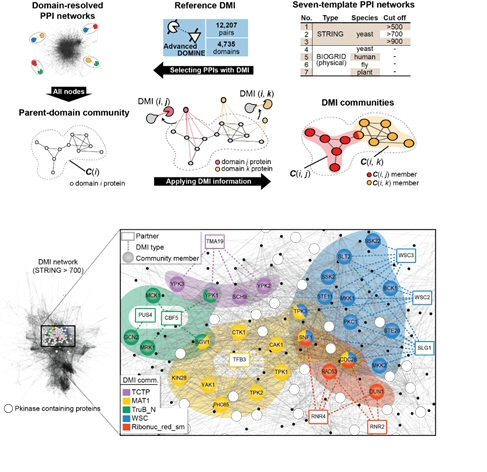
Figure 4. Construction of DMI community and network view of example DMI community.
IV. Evolutionary analysis of phenotypic divergence between human and model organism
Quantitative phenotype comparison of human and mouse orthologous genes
Systematic genetic screening of model organisms has become a crucial tool for understanding human gene-phenotype relationships. Model organisms, like mice, have been used to reveal phenotypic outcomes related to the manipulation of human orthologous genes to investigate human diseases. However, frequent dissimilarities of phenotypic outcomes across species, even when the ortholgous genes were perturbed, were constant issue for mouse genetic researchers. Dr. Kim’s group devised a method, which we named as Phenotype Similarity (PS) score, for the systematic quantification of phenotypic differences in human and mouse orthologous genes. Consequently, we compiled 642 HPGs (High Phenotype-similarity Genes) and 642 LPGs (Low Phenotype-similarity Genes) based on the PS score. [Figure 6]
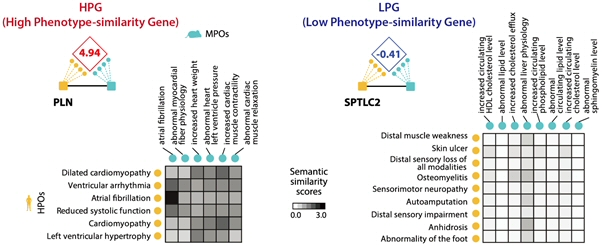
Han SK et al., 2018, MBE
Figure 5. Phenotype similarity (PS) score of orthologous human and mouse genes. Example of high PS score gene(HPG), PLN and low PS score gene(LPG), SPTLC2.
Correlation between divergence of noncoding regulatory sequence and phenotypic differences
Phenotypic differences have been suggested as the result of changes in noncoding regulatory sequences, which are frequently observed within mammalian species. Dr. Kim’s group compared the noncoding regulatory sequences of LPGs and HPGs, and found that LPGs included a greater proportion of significant sequence differences than HPGs. [Figure 7] Changes in gene regulatory sequences could trigger divergence in transcription across species. In this regard, we analyzed transcriptional divergence in orthologous genes with phenotypic differences and discovered that, in many cases, phenotypic differences can be significantly explained by transcriptome divergence across species. [Figure 7]
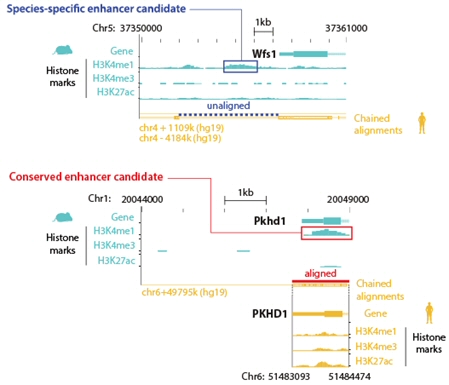
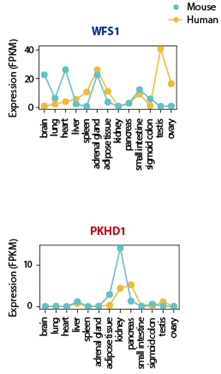
Han SK et al., 2018, MBE
Figure 7. Comparison of the conservation of noncoding regulatory sequence and expression pattern between LPG and HPG.
Modular approaches for the phenotypic changes of orthologous genes
Multiple genes rather than single one configure a phenotype, i.e., polygenicity, underlying disease module hypothesis and prioritization of disease gene candidates. Dr. Kim’s group analyzed the relationship between cross-species expression divergence of neighboring genes in functional modules and phenotypic discrepancies of human and mouse orthologs. We found certain gene and neighbors, connected by same bioprocess and pathway within either human or mouse, tightly associate between the phenotypic divergences of the gene and neighbors’ expression divergences.
